
José Samaniego Burneo 1 A; Santiago Guzmán García 2 B; Fabiana Samaniego Burneo 3 C; Eduardo Córdova Guzmán 4 D
- Médico General, Universidad Técnica Particular de Loja. Loja-Ecuador
- Reumatólogo. Departamento de Reumatología. Hospital Clínica San Agustín. Loja-Ecuador
- Maestra en Infectómica y Patogénesis Molecular. CINVESTAV. Ciudad de México-México
- Rotante Externo; consulta de Reumatología y Urología. Hospital Clínica San Agustín. Loja-Ecuador
A. ![]() ORCID: 0009-0004-0804-1170
ORCID: 0009-0004-0804-1170
B. ![]() ORCID: 0000-0002-1237-7927
ORCID: 0000-0002-1237-7927
C. ![]() ORCID: 0009-0008-1786-6093
ORCID: 0009-0008-1786-6093
D. ![]() ORCID: 0009-0006-5955-3672
ORCID: 0009-0006-5955-3672
Resumen
La linfohistiocitosis hemofagocítica es un padecimiento caracterizado por hiperactivación de los macrófagos y linfocitos T CD8+ e infiltración de estos en distintos órganos, lo que conlleva a un estado inflamatorio y muerte celular. Puede ser un trastorno primario o secundario de enfermedades autoinmunes, cáncer o infecciones. Es una enfermedad inusual pero frecuentemente fatal, que debe ser sospechada en pacientes con citopenia inexplicada con fiebre, y aunque se dispone de criterios valorativos, la poca especificidad y variedad de ellos desencadena un reto diagnóstico importante. A continuación, se presentará un caso clínico de un paciente de sexo masculino, que acude al servicio de urgencias por fiebre en estudio.
Abstract
The haemophagocytic lymphohistiocytosis is a clinicopathologic entity characterized by the macrophage´s and lymphocyte’s T CD8 hyperactivation and infiltration in many organs from the body, causing an inflammatory condition triggering cellular death. It can be a primary disease or secondary to malignancy, infection o autoimmune disease. The Hemophagocytic Syndrome is uncommon but it is usually lethal, and it should be considered in a patient with fever and unexplained cytopenias; it has been established diagnostic criteria, although the low specificity makes a diagnostic challenge. Following will be presented a clinical case of a male patient who comes to the emergency department for study of fever.
Introducción
La linfohistiocitosis hemofagocítica (LHH), también llamada síndrome hemafagocítico, es un trastorno hiperinflamatorio potencialmente mortal, que se debe a una hiperestimulación inflamatoria. Esta dolencia puede ser ocasionada por defectos congénitos o ser secundaria a múltiples enfermedades, siendo la forma autosómica recesiva la que confiere una mayor tasa de mortalidad (1). A nivel mundial, se tiene registro de 1.2 casos por cada millón de habitantes y tasas de mortalidad del 20 al 30% dentro de los dos meses iniciales del diagnóstico; sin embargo, se considera que esta cifra está subestimada, debido a la dificultad para realizar el diagnóstico (2,1). Por otro lado, aun cuando la LHH secundaria es más frecuente en niños y adultos jóvenes, es el tipo predominante en adultos mayores, aunque su prevalencia exacta no está bien establecida (3).
Clínicamente se caracteriza por fiebre y esplenomegalia acompañados de alteraciones bioquímicas, como citopenias, hipertrigliceridemia e hiperferritinemia, así como la demostración de hemofagocitosis en médula ósea, bazo o ganglios linfáticos (4, 5). A pesar de que se han formulado una serie de criterios para estandarizar el diagnóstico de la enfermedad, la poca especificidad de los síntomas y parámetros bioquímicos, aunado a la poca incidencia de la enfermedad, provocan un retraso en la valoración, que conlleva a una alta mortalidad (6).
Actualmente, hay una serie de esquemas de tratamientos con inmunosupresores y quimioterapia, que reduce significativamente la mortalidad, sobre todo si se inicia en las etapas tempranas (7).
Presentación del caso
Paciente masculino de 37 años de edad, con único antecedente patológico personal de litiasis renoureteral. Una semana antes de su ingreso, inició con un cuadro clínico caracterizado por cefalea de predominio frontal, opresiva, descrita como la peor de su vida, sin datos de focalización neurológica, además de dos picos febriles no cuantificados asociados a diaforesis profusa de predominio nocturno; por parte de medicina general, recibió tratamiento con cefixima por cuatro días, paracetamol e ibuprofeno sin mejoría, debido a lo cual acudió a urgencias. Al interrogatorio dirigido, negó viajes en los últimos tres meses, presenta síntomas cardiorrespiratorios, gastrointestinales, genitourinarios y dermatológicos, por lo que se administró analgésicos para control del dolor. Al examen físico, destacó la presencia adenopatías múltiples en cadena cervical anterior de manera bilateral, pequeñas, móviles, no dolorosas a la palpación, además de hepatomegalia de 2 cm por debajo del reborde costal y esplenomegalia grado 2; se realizó tomografía simple de cráneo, sin alteración; se tomó laboratorios en los que destacó leucopenia y un patrón colestásico en las pruebas de función hepática; se tomó hemocultivos sin desarrollo de microrganismos; se practicó punción lumbar en la que no hubo anormalidades en el citoquímico del líquido cefalorraquídeo (tabla 1), y se decidió su ingreso para continuar abordaje diagnóstico y terapéutico.
Durante su hospitalización, se interconsultó a infectología, y se inició tratamiento con ceftriaxona ante la sospecha de fiebre de etiología infecciosa; además, se solicitaron pruebas de influenza, cultivo en sangre, cultivos de secreción respiratoria, urocultivo, reacciones febriles, reacciones en cadena de polimerasa para micobacterias y virus, anticuerpos contra virus, tinción de Gram y cultivo de LCR, y fueron todas negativas, con lo que se descartó etiología infecciosa. A pesar del tratamiento antibiótico, continuó con cefalea y picos febriles; se tomó nuevos exámenes de laboratorio (tabla 2) en los que destacó posteriormente pancitopenia; se interconsultó a hematología; se realizó aspirado de médula ósea, en el cual se observó una médula ósea hipercelular (celularidad del 80%) con necrosis focal; detención de la diferenciación eosinofílica en la serie mieloide; no se observan microorganismos patógenos, únicamente llama la atención la presencia de una célula con características morfológicas de célula de Reed Sternberg. Luego se complementó con un PET CT con resultado de metabolismo glucolítico anormal en ganglios linfáticos mediastinales, hepatoesplenomegalia con inversión de la actividad hígado/bazo, porción medular del esqueleto axial y apendicular (figura 1 y 2); se tomó laboratorios complementarios y, al cumplir criterios para síndrome hemofagocítico (fiebre, esplenomegalia, citopenia periférica, hipertrigliceridemia, ferritina >500), se inició terapia con dexametasona 16 mg cada 24 horas.
Con el propósito de descartar causas secundarias de linfohistiocitosis hemofagocítica, se complementó con biopsia de ganglio linfático mediastínico y biopsia de hígado ecoguiada con resultados negativos para patologías secundarias; en la patología de ganglio linfático mediastinal, destacó macrófago con células sanguíneas intracitoplasmáticas, compatible con hemofagocitosis, que corroboró el diagnóstico de linfohistiocitosis hemofagocítica. Así mismo, fue valorado por reumatología, y después de complementar estudio de especificidades de antinucleares y complemento, se descartó causas autoinmunes. Además, se realizó broncoscopía con el objetivo de tomar cultivos de secreción respiratoria, realizar cepillado bronquial y lavado bronquial, donde no se identificaron células malignas, ni organismos patógenos. Al no tener una etiología secundaria, se decidió programarlo para esplenectomía en la que se descarta neoplasia, por lo que se concluyó el diagnóstico de linfohistociatosis hemofagocítica primaria, basándose en los criterios diagnósticos de la Sociedad de Histiocyte (7), y se inició tratamiento con etopósido con dexametasona, recibiendo ocho ciclos del mismo con adecuada respuesta clínica y bioquímica, sin presentar recurrencia de los síntomas.
Discusión
La linfohistiocitosis hemofagocítica es una enfermedad agresiva que compromete la vida. Aquí se produce una estimulación inmune excesiva pero ineficaz e implica grandes dificultades diagnósticas y terapéuticas. La incidencia se estima alrededor de 1.2 casos por millón de personas por año, pero seguramente se encuentra subestimada. La linfohistiocitosis hemofagocítica comprende dos condiciones diferentes que pueden ser difíciles de distinguir una de la otra: una forma primaria o familiar y una forma secundaria o adquirida, provocadas por una variedad de eventos que alteran la homeostasis inmune. La forma primaria, genética o familiar, se refiere a la linfohistiocitosis hemofagocítica causada por la mutación de un gen. La forma secundaria, esporádica o adquirida se refiere a la linfohistiocitosis hemofagocítica en la que se ha identificado un claro desencadenante. Las infecciones son un desencadenante común en aquellas personas con una predisposición genética y en los casos esporádicos (8, 9).
La linfohistiocitosis hemofagocítica primaria o familiar, con patrón de herencia autosómica recesiva, es una enfermedad mortal con una supervivencia media de menos de 2 meses después del diagnóstico si no se trata. Por lo general, tiene su inicio durante la infancia. Las pruebas genéticas han demostrado que la forma primaria puede ocurrir a cualquier edad, desde la presentación en útero con hidropesía fetal hasta tan tarde como los 70 años de edad. La linfohistiocitosis hemofagocítica secundaria se desarrolla como resultado de la fuerte activación del sistema inmune, que puede ser desencadenada por una infección grave (49%), tumores malignos (27%), enfermedades autoinmunes (7%), algunas enfermedades metabólicas y síndromes de deficiencia inmune (6%). Es importante destacar que el inicio y los brotes de la enfermedad, tanto primaria como secundaria, pueden ser desencadenados por infecciones (10).
A pesar de los avances en el conocimiento, la patogenia de la linfohistiocitosis hemofagocítica aún no está clara. Desde la primera descripción de las mutaciones del gen perforina, emitida por Stepp en 1999, ha surgido información valiosa sobre las mutaciones genéticas que dan lugar al fenotipo de la linfohistiocitosis hemofagocítica. En la actualidad, del 20 al 50% de los genes implicados en la patogenia se desconocen; lo que está claro es que todas las anomalías genéticas conocidas conducen a defectos en proteínas que desempeñan un papel importante durante la secreción en la vía citolítica (11).
La forma familiar o primaria se cree que se debe a la terminación defectuosa de la respuesta inmune, que resulta en la activación persistente de los macrófagos y las células T citotóxicas. Una hipótesis alternativa implica el fracaso para eliminar antígenos que resultan en la estimulación continua de las células efectoras inmunes. Es posible que tanto el fracaso para eliminar el antígeno que provoca una estimulación continua y el fracaso para terminar la respuesta inmune jueguen un papel importante. La patogenia de la forma secundaria o adquirida es aún menos clara, aunque se ha encontrado que estos pacientes tienen cambios heterocigotos o polimorfismos en los genes de la linfohistiocitosis hemofagocítica primaria.
Aunque la patogenia exacta de la enfermedad no se conoce bien, es evidente que las manifestaciones clínicas se deben a la hiperactivación de los linfocitos T CD8 y los macrófagos; la proliferación, la migración y la infiltración de estas células en varios órganos, y a la hiperproducción de citocinas con niveles persistentemente elevados de las múltiples citocinas proinflamatorias en sangre, lo que resulta en disfunción orgánica progresiva que puede conducir a la muerte (3).
El problema primario al diagnóstico de la linfohistiocitosis hemofagocítica es que carece de marcadores específicos de la enfermedad; la clínica es inespecífica y es caracterizada, independientemente de la etiología, por fiebre, hepatoesplenomegalia, citopenias y el hallazgo de macrófagos activados en órganos hematopoyéticos, por lo que resulta en ocasiones en falla orgánica y muerte (12). Igualmente, pueden coexistir con manifestaciones neurológicas, resultado de la infiltración de macrófagos, y con mal pronóstico cuando llegan a presentarse, y manifestaciones dermatológicas, tales como eritrodermia, rash maculopapular generalizado, petequias y púrpura.
Los exámenes de laboratorio dirigidos a evaluar el síndrome se realizan en el abordaje de una fiebre de origen desconocido y consisten en biometrías hemáticas, estudios de coagulación y pruebas de función hepática, y cuando se tiene la sospecha, se agrega adicionales marcadores, como ferritina (>500 mg/dL), triglicéridos y estudios inmunológicos. Adicionalmente, aunque no se ha comprobado la utilidad clínica, se recomienda solicitar las mutaciones puntuales de dicha patología (PRF1, UNC13D, STX11 y UNC18B), así como otros paraclínicos que son de utilidad, como hipoalbuminemia, DHL, Dímero D y PCR elevado, cultivos, serologías o PETCT en caso de sospecha de un factor desencadenante (13, 14).
En el 2004, se propusieron criterios diagnósticos que definieran el síndrome, los cuales se resumen en dos puntos: A) Diagnóstico molecular (mutaciones genéticas puntuales PRF1, UNC13D, STXVP1, SH2D1A o XIAP), y B) Cinco criterios clínicos (fiebre, esplenomegalia, citopenias periféricas (mínimo 2); hipertrigliceridemia y/o hipofibrinogenemia; hemofagocitosis en la médula ósea, bazo, ganglios linfáticos o hígado (la modalidad de elección es la biopsia de médula ósea por su accesibilidad y menor complicación); ferritina >500 ug/L; baja o ausente actividad de las células NK, e incremento de la concentración de CD25 >2400 U/Ml5. Como factores de mal pronóstico, se tienen en cuenta la hipoalbuminemia, trombocitopenia e hiperferritinemia (>50,000) pues incrementan la mortalidad. Deben cumplirse 5 de los 8 criterios para realizar el diagnóstico, y son los que se utilizan actualmente; puede que no todos los pacientes cumplan los 5 criterios, por lo que debe evaluarse caso por caso (7, 15).
Por otra parte, aunque una elevación de ferritina es un indicador altamente sensible y específico para el síndrome hemofagocítico (SHF) en niños, su especificidad disminuye en adultos, y no es un buen predictor de la enfermedad en esta población. No obstante, un nivel elevado de ferritina siempre debe llevar a considerar la posibilidad de SHF. Los estudios genéticos y funcionales no se recomiendan en adultos debido a que las anomalías son raramente detectables. El “Hscore” resulta ser una herramienta útil para un SHF reactivo, ya que estima la probabilidad en función de variables clínicas, biológicas y citológicas (7; 12).
Tabla 1. Criterios diagnósticos de la Sociedad de Histiocyte

Fuente: Otárola B. et al., 2020 (7).
Un tratamiento oportuno es esencial para la supervivencia de los pacientes afectados; con frecuencia, la barrera más común para un resultado exitoso es un retraso en el diagnóstico, el cual es difícil debido a la rareza de este síndrome, la presentación clínica variable, la falta de especificidad de los hallazgos clínicos y de laboratorio. Antes de la iniciación de los regímenes terapéuticos actuales, la tasa de supervivencia a 1 año de los pacientes con linfohistiocitosis hemofagocítico primaria estaba cerca del 0% (6). El tratamiento actual en los adultos está centrado en la supresión del estado hiperinflamatorio, enfocado en la destrucción de linfocitos T citotóxicos y macrófagos con citotóxicos, terapia inmunosupresora y el tratamiento de cualquier etiología desencadenadora del síndrome hemofagocítico.
La terapéutica está basada en el protocolo pediátrico HLH-94/-2004, considerando un régimen de 8 semanas, con un curso de inducción de dexametasona, etopósido, ciclosporina y, en caso de manifestaciones neurológicas, metrotexate intratecal e hidrocortisona. Sin embargo, muchos hematólogos prefieren como terapia adicional por persistencia de la enfermedad a la ciclosporina o tacrolimus; este último es el de elección por su menor nefrotoxicidad (2). Cuando se tiene una causa identificable responsable del inicio del síndrome hemofagocítico, tal como un proceso infeccioso, y el paciente está estable, puede responder a la terapia dirigida contra el microorganismo; no obstante, si existe deterioro clínico, sería necesario iniciar la quimioterapia antes descrita, exceptuando infección por Epstein Barr, que requiere terapia con etopósido coadyuvante.
Las causas inmunológicas requerirán tratamiento con inmunosupresores a dosis altas y, cuando se trata de las malignas, es necesario una quimioterapia dirigida al proceso oncológico (4). En el caso de que no se encuentre el desencadenante, debería realizarse un trasplante alogénico de médula ósea, pero evitándose cuando existe actividad por alto riesgo de rechazo o iniciar terapia con alemtuzumab para atenuar la enfermedad (8). El primer protocolo terapéutico, organizado en 1994, resultó en una tasa de supervivencia global del 55%, con una probabilidad de supervivencia del 62% para los pacientes manejados con trasplante de células hematopoyéticas. Posteriormente, con la introducción del acondicionamiento de intensidad reducida y una mayor experiencia, la supervivencia después del trasplante ha mejorado hasta un 92%. Las tasas de mortalidad reportadas en la linfohistiocitosis hemofagocítica secundaria varían del 8 al 22%. El retraso en el diagnóstico y la afectación multiorgánica se asocian con un pronóstico inferior, ya sea en la forma primaria o secundaria, y la terapéutica necesita ser establecida lo antes posible para prevenir el daño tisular irreversible (6; 11).
Conclusiones
En resumen, la linfohistiocitosis hemofagocítica es una enfermedad rara, poco sospechada en la práctica clínica, en la que el diagnóstico suele realizarse de manera tardía por la variabilidad de presentación clínica y las diferentes etiologías. Es importante considerarlo en los pacientes con fiebre de origen desconocido y citopenias de etiología indeterminada para brindar un tratamiento oportuno, disminuir las complicaciones y la mortalidad, y siempre, ante estos casos, se necesita un abordaje multidisciplinario para tomar decisiones en conjunto.
Financiamiento
Los autores no recibieron financiación para el desarrollo de la presente investigación.
Conflictos de interés
Los autores declaran que no existe conflicto de interés.
Tabla 2. Resumen de estudios de laboratorio

Imágenes
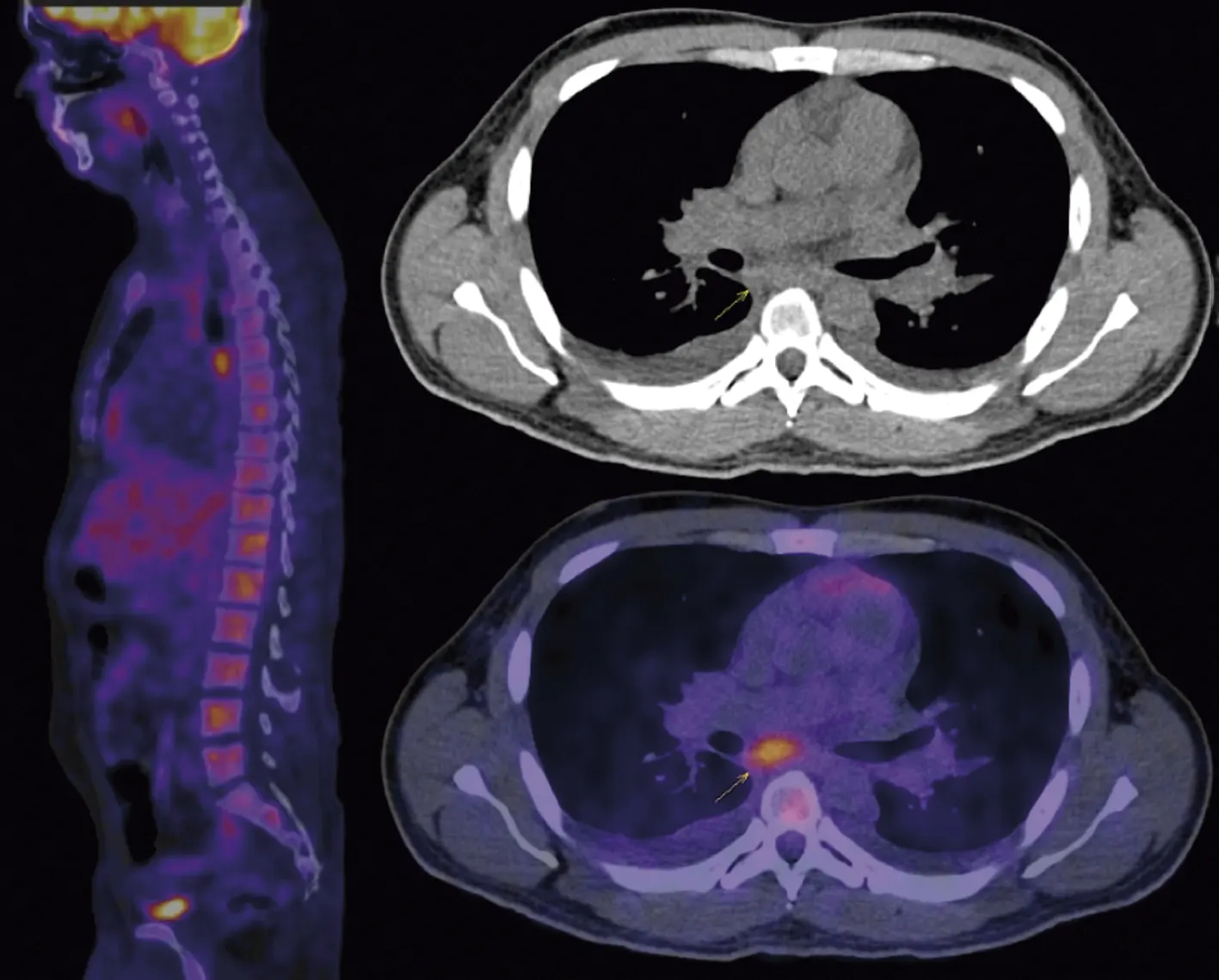
Figura 1.
PET-CT con 18 F-FDG. Aumento de la actividad glucolítica en la porción medular del esqueleto axial, así como en un ganglio subcarinal de 29 mm y otro mediastinal. Existe concentración fisiológica del radiofármaco en cerebro, anillo de Waldeyer, miocardio, hígado, intestino y vejiga.
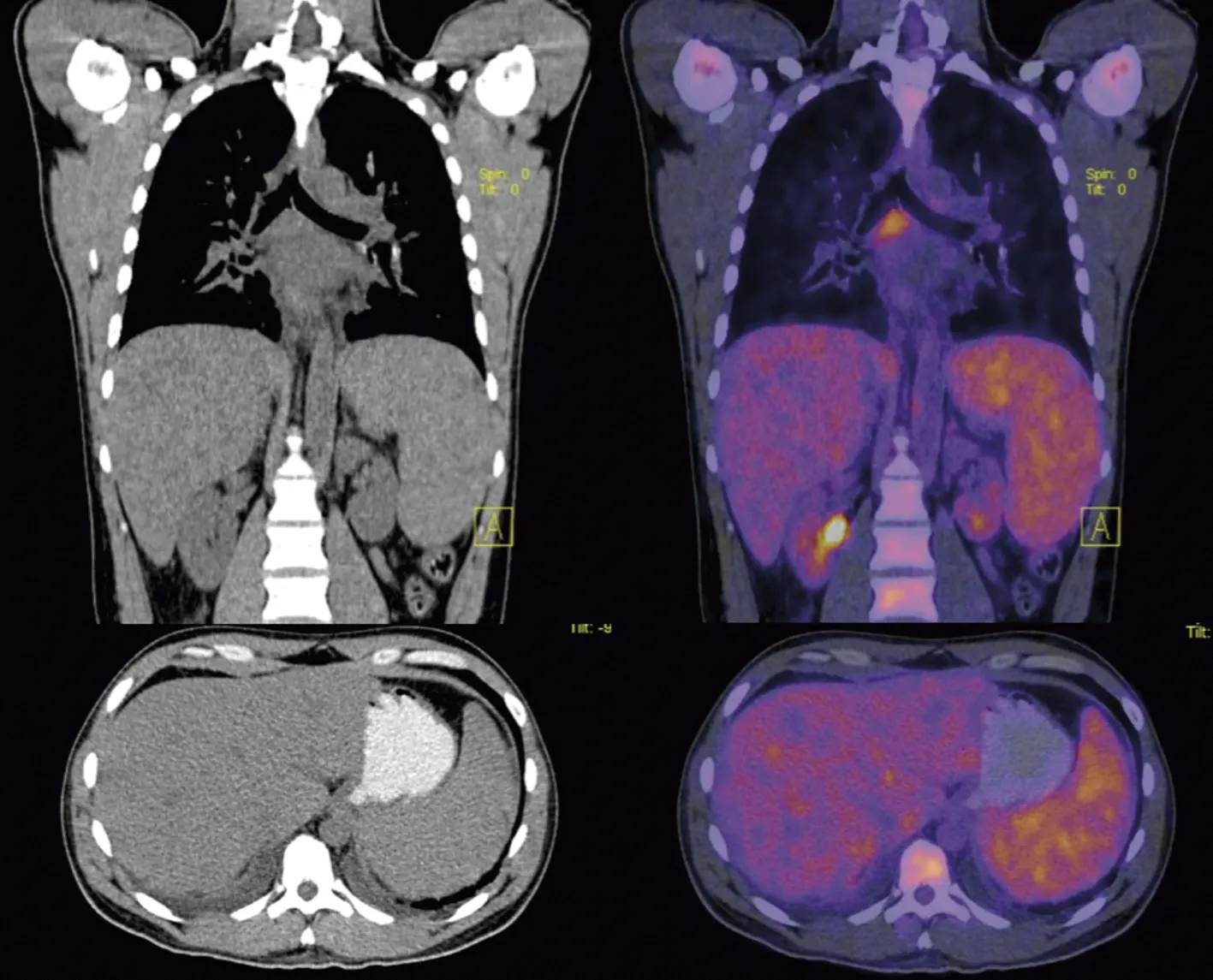
Figura 2.
PET-CT con 18 F-FDG. Metabolismo glucolítico anormal en ganglio subcarinal y hepatoesplenomegalia con inversión de la relación de actividad hígado-bazo. Existe concentración fisiológica del radiofármaco en riñones.