

Franklin Hidalgo-Coronel 1 A; Julia Jaramillo-Requena 1 B; Alejandra Cango-Calderon 1 C; Natalia Bailon-Moscoso 1, 2 D
- Carrera de Medicina, Universidad Técnica Particular de Loja, Ecuador. San Cayetano alto s/n. CP 1101608
- Departamento de Ciencias de la Salud, Universidad Técnica Particular de Loja, Ecuador. San Cayetano alto s/n. CP 1101608
A. ![]() ORCID: 0009-0009-6710-5695
ORCID: 0009-0009-6710-5695
B. ![]() ORCID: 0009-0006-7809-693X
ORCID: 0009-0006-7809-693X
C. ![]() ORCID: 0009-0007-9954-9595
ORCID: 0009-0007-9954-9595
D. ![]() ORCID: 0000-0003-2754-1328
ORCID: 0000-0003-2754-1328
Resumen
El síndrome de Hermansky-Pudlak (SPH) es un trastorno genético autosómico recesivo, que cursa con albinismo oculocutáneo, disfunción plaquetaria, colitis granulomatosa y fibrosis pulmonar. Los individuos que padecen de este síndrome pueden presentar alteraciones en 1 de 11 genes implicados (AP3B1, AP3D1, BLOC1S3, BLOC1S5, BLOC1S6, DTNBP1, SHP1, SHP3, SHP4, SHP5 o SHP6). Aunque este síndrome es frecuente en la población puertorriqueña, grupo demográfico en el que los subtipos más frecuentes son el SHP-1 y el SHP-3, sin embargo, se han reportado casos de esta patología a escala mundial. Los mecanismos fisiopatológicos implicados son variados, pero el principal problema radica en las alteraciones que los genes ya mencionados producen en la creación y el tráfico de orgánulos relacionados con lisosomas. Los dos rasgos más característicos son la diátesis hemorrágica por ausencia de los gránulos densos en las plaquetas y el albinismo oculocutáneo por ausencia de melanosomas en las células melanocíticas. El diagnóstico de esta patología se basa en los hallazgos clínicos (albinismo oculocutáneo y diátesis hemorrágica), además de la microscopía electrónica para determinar la ausencia de gránulos densos en plaquetas. El diagnóstico también puede ser establecido mediante pruebas genéticas, y sobre todo estas son útiles para determinar el subtipo específico del SHP. El tratamiento incluye medidas preventivas para el cuidado de piel, ojos y pulmones; en caso de colitis, se aconsejan fármacos como los corticoides o rituximab.
Abstract
Hermansky-Pudlak syndrome is an autosomal recessive genetic disorder that causes oculocutaneous albinism, platelet dysfunction, granulomatous colitis, and pulmonary fibrosis. Individuals with this syndrome may have alterations in 1 of 11 genes involved (AP3B1, AP3D1, BLOC1S3, BLOC1S5, BLOC1S6, DTNBP1, SHP1, SHP3, SHP4, SHP5 or SHP6). This syndrome is common in the Puerto Rican population, and generally within this demographic group, the most common subtypes are HPS-1 and HPS-3; however, cases of this pathology have been reported worldwide. The pathophysiological mechanisms involved are varied, but the main problem is the alterations that the genes previously mentioned produces on the creation and trafficking of lysosome-related organelles, being the two most characteristic features hemorrhagic diathesis due to the absence of dense granules in platelets and oculocutaneous albinism, due to the absence of melanosomes in melanocytic cells. The diagnosis of this pathology is based on clinical findings (oculocutaneous albinism and hemorrhagic diathesis), in addition to electron microscopy to determine the absence of dense granules in platelets. The diagnosis can also be established by genetic tests, and these are especially useful to determine the specific subtype of HPS. Treatment includes preventive measures for the care of skin, eyes and lungs; in case of colitis, drugs such as corticosteroids or rituximab are recommended.
Introducción
El síndrome de Hermansky-Pudlak (SHP) es una patología de características autosómicas recesivas y, además, es multisistémica, puesto que es responsable de generar alteraciones en distintos órganos y sistemas de quien la padece [1]. Este trastorno genético posee 11 subtipos, cada uno establecido por la presencia de variantes patogénicas genéticas en 1 de 11 genes posibles pertenecientes a 4 subunidades proteicas (AP-3, BLOC-1, BLOC-2, BLOC-3), estas subunidades comprenden un total de 11 genes los cuáles se pueden ver alterados en esta enfermedad (AP3B1, AP3D1, BLOC1S3, BLOC1S5, BLOC1S6, DTNBP1, SHP1, SHP3, SHP4, SHP5 o SHP6) [2,3]. El descubrimiento de esta patología se dio gracias a los médicos checoslovacos Frantisek Hermansky y Paulus Pudlak, quienes encontraron esta enfermedad en dos individuos que presentaban albinismo oculocutáneo acompañado de trastornos hemorrágicos [3]. La sintomatología de esta enfermedad genética rara es variada; generalmente se manifiesta con albinismo oculocutáneo, disfunción plaquetaria por alteraciones en la agregación plaquetaria, colitis granulomatosa y fibrosis pulmonar (figura 1) [4]. No obstante, no en todos los individuos se presentarán exactamente las mismas manifestaciones clínicas, sino que estarán sujetas al gen defectuoso en cuestión [2]. Por lo común, en estos pacientes la principal causa de muerte es atribuida a la fibrosis pulmonar; la edad de aparición de este trastorno pulmonar en los pacientes con SHP es incluso a una edad más temprana que los pacientes que padecen de fibrosis pulmonar idiopática, por lo cual la esperanza de vida disminuye considerablemente y se ubica entre los 30 y los 50 años [4,5]. El presente ensayo intenta realizar una revisión con bibliografía reciente de los mecanismos fisiopatológicos y genéticos que intervienen en el desarrollo de esta enfermedad autosómica recesiva y el porqué de las principales complicaciones, que pueden disminuir la calidad de vida de estos pacientes.
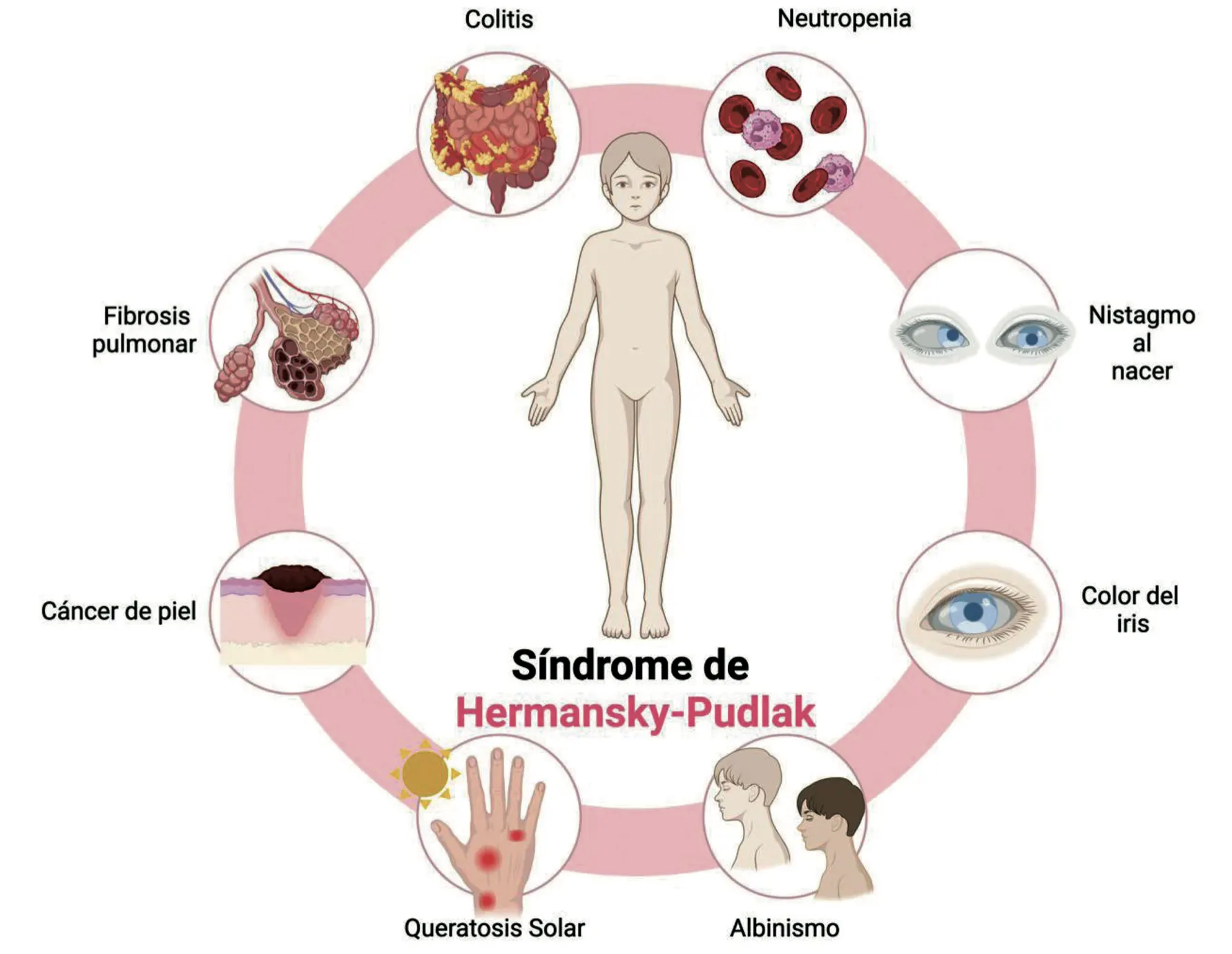
Figura 1. Manifestaciones clínicas generales del síndrome de Hermansky-Pudlak
Epidemiología
La aparición del SHP es inusual y afecta a alrededor de 1 a 9 personas por cada millón de habitantes [3]. Sin embargo, la presencia de este trastorno genético en el país de Puerto Rico es bastante frecuente, se estima que 1 de cada 1800 puertorriqueños se ven afectados, existiendo una tasa de portadores de 1:21; en este país los subtipos del SHP más comunes son el SHP-1 y el SHP-3, que cursan con afección colónica y pulmonar variable [6].
No obstante, la extensión del SHP va más allá de Puerto Rico, dada la mutabilidad genética que posee este síndrome al presentar 11 variables, en la literatura se han reportado múltiples casos que afectan más allá del país centroamericano. Por ejemplo, si bien el SHP-1 es frecuente en el noroeste de Puerto Rico, este subtipo también se ha visto en un pueblo suizo y, como un aislamiento genético, en Japón [7]. El SHP-2 se ha reportado muy poco en la literatura; a pesar de ello, se ha llegado a evidenciar en 3 de 8 pacientes poseedores del síndrome de Hermansky-Pudlak en Omán [8] y también existe un caso reportado en China [9], el SHP-6 también ha sido reportado en estos mismos países [8,9].
Por su parte, el SHP-3 es frecuente en el centro de Puerto Rico, pero, además, se han reportado casos a escala mundial en China, Medio Oriente, América del Sur y Europa occidental y oriental [7,9]. Los subtipos SHP-4, SHP-5 (e incluso el SHP-1) se han llegado a reportar incluso en pacientes con ascendencia mexicana, uruguaya, hondureña, cubana, venezolana y salvadoreña [10]. El SHP-7 ha llegado a ser descrito en Paraguay [11]. El SHP-8 se describió en 2006 en pacientes pakistanís configurando el descubrimiento de esta nueva variante [12]. El SHP-9 ha sido escasamente reportado, sin embargo, la literatura ha reportado un caso en Irán [13]. En Turquía se reportó por primera vez un caso del SHP-10 en un paciente que era hijo de padres turcos, que eran primos hermanos [14], mientras que el SHP-11 ha sido reportado en una familia consanguínea de China [15].
Toda esta evidencia epidemiológica no hace más que reforzar la conjetura de que el síndrome de Hermansky-Pudlak es una enfermedad de escala mundial que ha sido reportada en un sinnúmero de países y no únicamente en Puerto Rico. La aparición de esta enfermedad varía no solo según el país, sino también según el subtipo específico que se produce en cada uno de ellos.
Manifestaciones clínicas y su mecanismo fisiopatológico
Fisiopatología
Es importante relacionar la patogenia con las manifestaciones clínicas que presentan de manera general. Para comprender mejor el tema, es preciso saber que, de los once genes actualmente conocidos, todos son componentes de complejos heterooligómeros involucrados en la producción y tráfico de orgánulos relacionados con lisosomas (LRO); , estos complejos son dos: 1) Complejo de biogénesis de orgánulos relacionados con lisosomas (BLOC-1, 2 y 3) y 2) Proteína adaptadora-3 (AP-3). Ambos son conocidos como complejos asociados a proteínas de SHP (HPAC).
Los LRO producidos y transportados a través de estos complejos son necesarios para la producción y función de melanosomas, gránulos delta de plaquetas, gránulos de linfocitos y cuerpos lamelares [16]. Por cada característica clínica, se presentan manifestaciones distintas relacionadas con los defectos en el LRO por la alteración en genes que actúan sobre los HPAC [17]. En la figura 2 se observa la vía endosómica trans-Golgi, la cual es fundamental en el desarrollo de los orgánulos relacionados con los lisosomas; dentro de este proceso, las GTPasas Rab32 y Rab38 son de vital importancia, ya que se encargan de regular el tráfico de vesículas de esta red, así como la maduración de los orgánulos relacionados con lisosomas [18]. Las alteraciones en los genes por el síndrome de Hermansky-Pudlak pueden afectar estas GTPasas, además de cualquier punto de la red endosómica trans-Golgi, ocasionando los problemas en el tráfico y maduración de los LROs [19], y produciendo la clínica característica de este síndrome que se detalla a continuación.
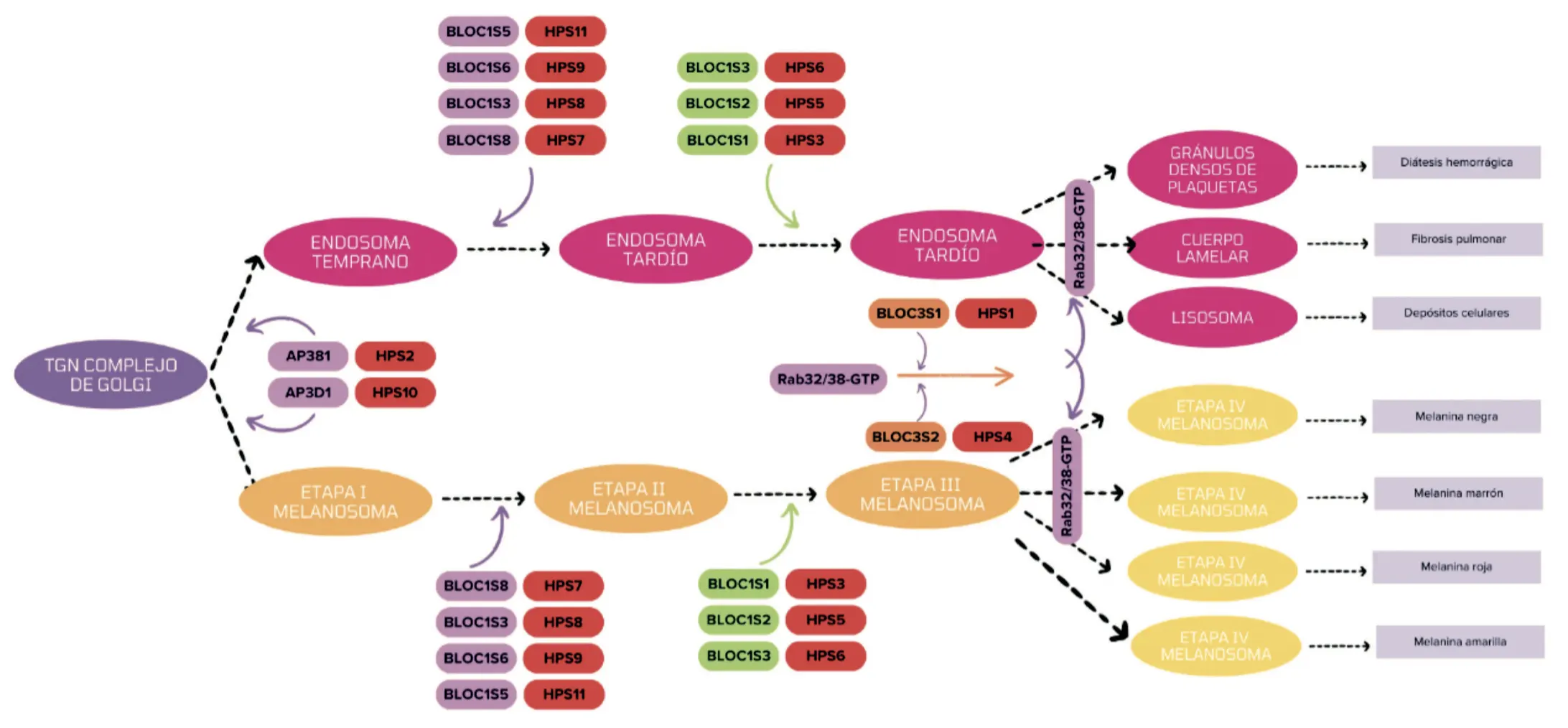
Figura 2. Vía endosómica de la red trans-Golgi. En la vía endosómica trans-Golgi, intervienen los endosomas tempranos los cuales son los compartimentos iniciales en donde se recibe el material internalizado desde la membrana plasmática, y actúan como centro de clasificación para saber si las moléculas son recicladas o trasportadas al aparato de Golgi o enviados hacia los endosomas tardíos para su degradación. Complejos como BLOC-1 o 2 son esenciales para el tráfico de proteínas desde los endosomas tempranos [20]. Por su parte, los endosomas tardíos surgen a partir de los endosomas tempranos, estos cuando maduran se fusionan con lisosomas para formar endolisosomas encargados de la degradación del contenido, o pueden seguir rutas específicas dirigidas por los complejos BLOC para la creación de LROs como los gránulos densos, los cuerpos lamelares [21]. En cuanto a la vía melanosomal, los complejos BLOC son cruciales para la salida selectiva de proteínas desde los endosomas tempranos hacia los melanosomas [21]. Cualquier alteración a nivel de los complejos BLOC puede producir la deficiencia o inmadurez de los LRO, conllevando a subtipos específicos de SHP.
Manifestaciones cutáneas
Según lo publicado por Li, W. [17], todos los subtipos del SHP tienen albinismo cutáneo relacionado con la deficiencia de melanosomas en los melanocitos de la piel cuando se producen mutaciones en los complejos AP-3, BLOC-1, -2, -3 [3], por lo cual los individuos manifiestan piel pálida y cabello claro o muy claro. Este factor deja expuestas a estas personas al desarrollo de lesiones cancerígenas en piel, lo que es un problema común en los afectados [22].
Así mismo, el albinismo ocular también está relacionado con la ausencia de melanosomas [23], células epiteliales pigmentarias de la retina que están presentes en todos los HPAC: AP-3, BLOC-1,-2,-3 [3], los cuales causan afección cutánea, correlacionando así el albinismo oculocutáneo típico de este síndrome. Esta alteración provoca nistagmo, reducción del pigmento del iris, reducción del pigmento de la retina, hipoplasia foveal con reducción significativa de la agudeza visual (normalmente en el rango de 20/50 a 20/400) y estrabismo en varios individuos [3,24].
Diátesis hemorrágica
Otra característica clínica importante de este síndrome es la diátesis hemorrágica, que se produce por disfunción plaquetaria. El LRO involucrado son los gránulos densos de las plaquetas [25] y el cuerpo de Weibel-Palade, ambos presentes en células endoteliales, causantes de los síntomas hemorrágicos del SHP [26]. Los gránulos densos plaquetarios contienen sustancias cruciales para la coagulación, como el ADP, ATP y calcio; su deficiencia es responsable de generar alteraciones en la agregación plaquetaria. En las personas con SHP, no se observa la presencia de los gránulos densos debido a la presencia de mutaciones genéticas en AP-3 [7], y en BLOC1 o BLOC2 o BLOC3, Estas 3 subunidades están implicadas, en la formación, fusión y tráfico de vesículas. Estas vesículas, son las que posteriormente se convertirán en gránulos densos, los cuáles son pequeños orgánulos secretores de diversas sustancias que permiten la agregación plaquetaria. En el síndrome de Hermansky Pudlak, las mutaciones producidas a nivel de estas subunidades, impedirán la adecuada biogénesis de los gránulos densos, lo que producirá alteraciones en la hemostasia primaria, provocando hematomas, epistaxis, hemorragia gingival, hemorragia después del parto, sangrado digestivo y sangrado prolongado en la menstruación o después de las cirugías [3].
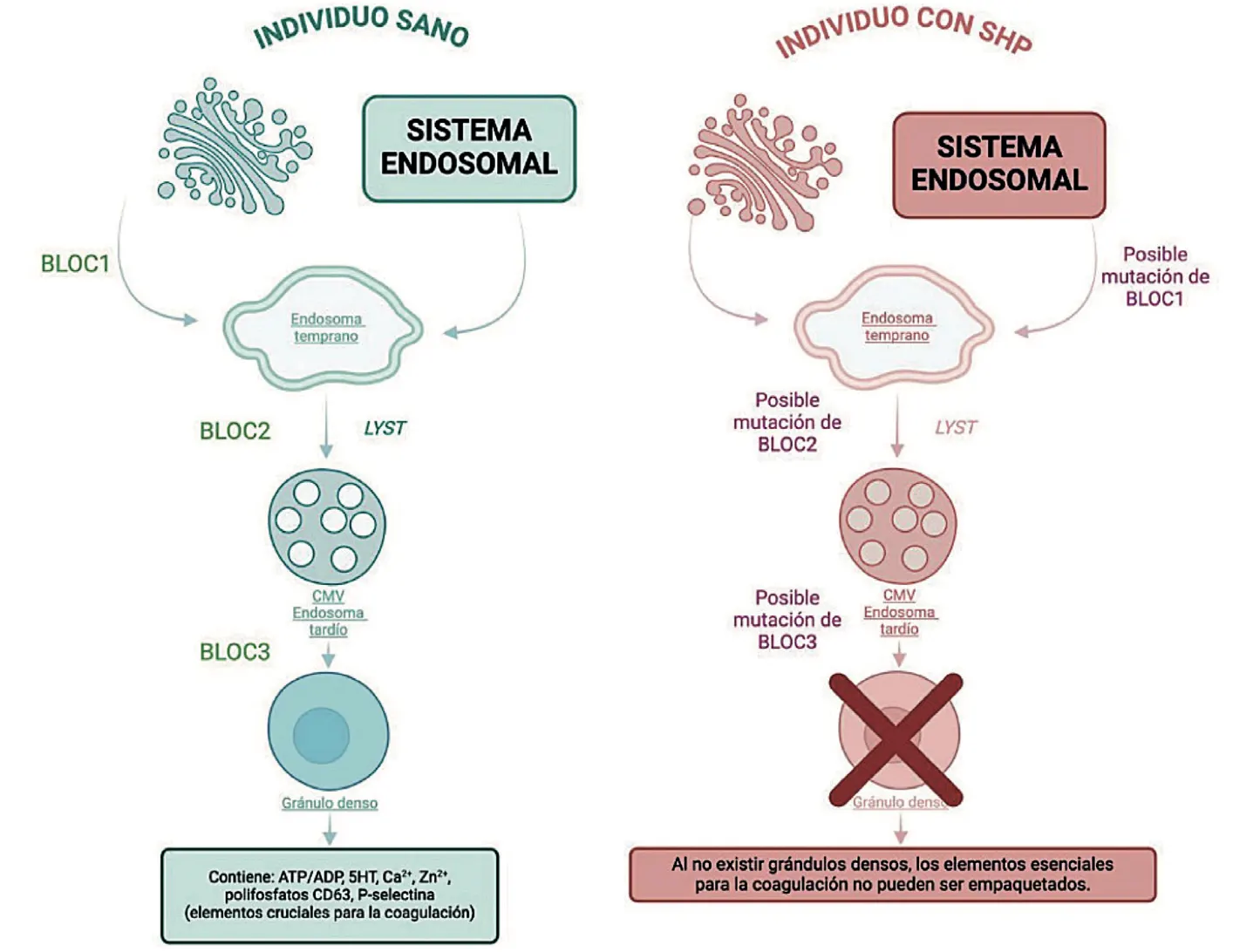
Figura 3. Fisiopatología de los trastornos de la coagulación en individuos con SHP. En un individuo sano, la red-trans-Golgi permite la creación de los gránulos densos gracias a los endosomas tempranos y tardíos, los cuales requieren de los complejos BLOC para la creación y maduración de estos LRO. Los gránulos densos son responsables de contener sustancias como el ADP, calcio y serotonina, que son moléculas cruciales para la agregación plaquetaria y la formación del trompo. En un individuo con síndrome de Hermansky-Pudlak, las mutaciones sobre BLOC1-2-3 pueden provocar varias alteraciones a nivel de este proceso, como la deficiencia de transporte de proteínas para la biogénesis de gránulos densos o la alteración en el contenido de los endosomas tempranos, lo cual produce en estos pacientes alteraciones irreversibles en la coagulación.
Fibrosis pulmonar
Un trastorno crucial que presentan los pacientes con SHP es la fibrosis pulmonar, en la que intervienen los cuerpos lamelares, que son las células epiteliales alveolares de tipo II, que contiene surfactante. En este caso, los HPAC involucrados son únicamente BLOC-3 y AP-3 [28]. La alteración en los genes de esta subunidad proteica provoca hidrogénesis progresiva del parénquima pulmonar y de los septos interalveolares, y el resultado final es la muerte por insuficiencia respiratoria. El cuadro clínico cursa con disnea e hipoxemia debilitante creciente, y afecta principalmente a niños y adultos en la cuarta o quinta década de vida [29,30].
La fibrosis pulmonar que se manifiesta en algunos subtipos de esta patología es un factor de mal pronóstico comparado con otros subtipos de SHP. La radiografía es una herramienta útil para el diagnóstico de esta alteración; no obstante, las alteraciones observadas pueden ser diversas e incluyen diminución del volumen pulmonar o panalización, bronquiectasias, aumento del grosor septal y pleural, opacidad reticular y patrón en vidrio esmerilado. Estos hallazgos radiográficos son generalmente observados en la parte periférica del pulmón y avanzan hasta la región central de los pulmones [30].
Enfermedad inflamatoria intestinal
Al igual que la fibrosis pulmonar, la enfermedad inflamatoria intestinal es una complicación derivada de la acumulación en múltiples órganos de un complejo lípido-proteico amorfo denominado lipofuscina ceroidea [26]. En referencia al tema, Li, W, et al. reportan que los lisosomas implicados en la enfermedad inflamatoria intestinal son las grandes vesículas de núcleo denso, las cuales son orgánulos secretores que se encuentran en ciertos tipos de células, incluidas las células de Paneth en el intestino y, a su vez, se relaciona con la mutación en el grupo completo BLOC [17]. En el más reciente estudio sobre la enfermedad inflamatoria intestinal en SHP, los resultados muestran que los síntomas se manifiestan inicialmente en niños, incluidos algunos de 5 años de edad o menos, y en adultos, el cuadro clínico fue hematoquecia, dolor abdominal, heces blandas y dolor abdominal tipo cólico acompañados de rectorragia, estreñimiento, tenesmo, náuseas y/o vómitos, fiebre, pérdida de peso, melena y sangrado gastrointestinal superior, pero no se encontró cáncer de colon [31].
Si bien el estudio de la enfermedad inflamatoria intestinal en los pacientes con SHP es una situación complicada, pues están altamente expuestos a la producción de hemorragias en el tracto digestivo por alteración plaquetaria como ya se ha descrito. La última evidencia manifiesta que la colitis producida en esta enfermedad no solo se relaciona con alteraciones lisosómicas y del sistema inmune innato, sino también al almacenamiento de lípidos y a la enfermedad metabólica subyacente, donde tanto los monocitos como los macrófagos un papel fundamental en la patogenia de esta alteración [32].
Neutropenia
La neutropenia, e incluso la neutropenia congénita, es una característica clínica importante de esta patología que se manifiesta con inmunodeficiencia relacionada con la falta de gránulos líticos y azurófilos en los neutrófilos. El transporte anormal de gránulos azurófilos conduce a una maduración celular deteriorada, lo que resulta en neutropenia [33]; la biogénesis y el contenido de estos orgánulos intracelulares afectarían la citotoxicidad de las células linfocitos T citotóxicos y los NK y la acumulación de proteína CD63 en la membrana plasmática en pacientes con complejo AP-3 defectuoso [34].
Todo esto, a su vez, se relaciona con la inmunodeficiencia donde el compartimento MHC-II es el LRO afectado [17], lo cual resulta en una clínica de inmunodeficiencia acompañada de infecciones virales y bacterianas recurrentes [34–36].
Consideraciones genéticas
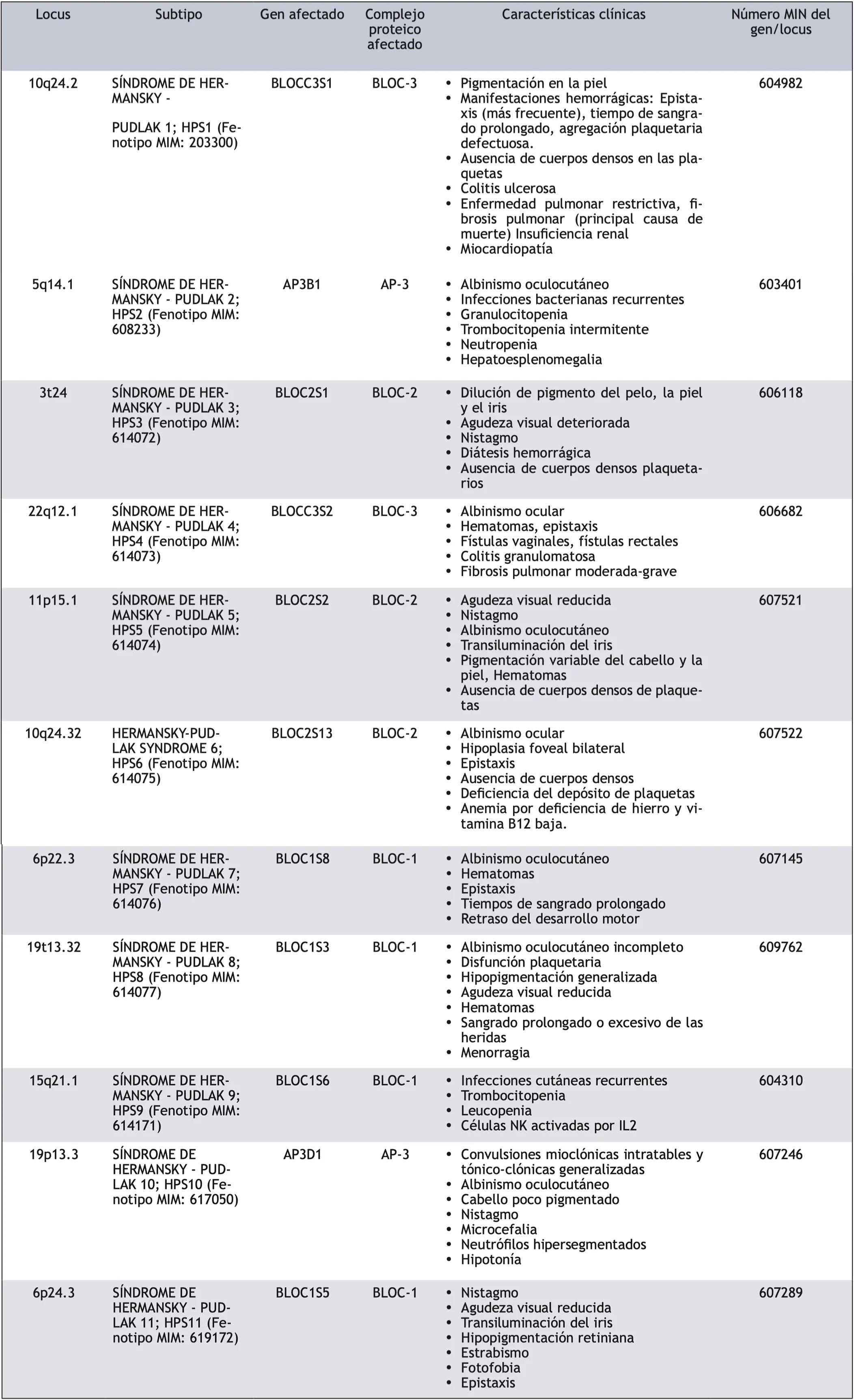
Esta patología autosómica recesiva está atribuida a la deficiencia de variantes patogénicas bialélicas de 11 genes humanos (AP3B1, AP3D1, BLOC1S3, BLOC1S5, BLOC1S6, DTNBP1, SHP1, SHP3, SHP4, SHP5 o SHP6). La deficiencia de cualquiera de estos 11 genes puede producir el SHP (tabla 1); el SHP-1 es el que aparece con más frecuencia en el 37.5% de los casos [7,37,38].
Tabla 1. Subtipo de Hermansky-Pudlak y gen y subunidad proteica afectada
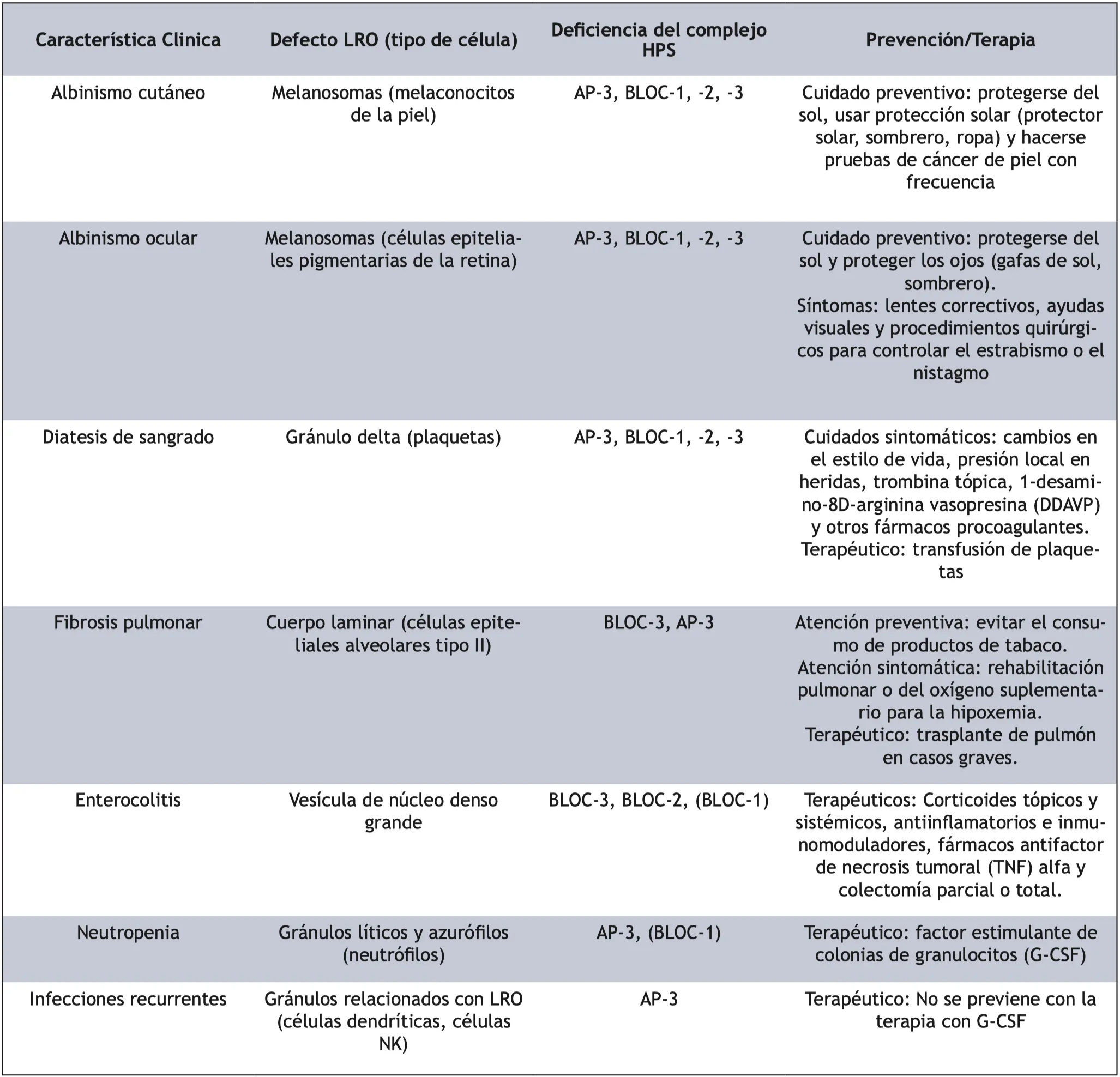
El síndrome de Hermansky Pudlak afecta sobre todo al conjunto de genes que se encargan de la síntesis de complejo de diversas subunidades como AP-3, BLOC1, BLOC2 y BLOC3; mutaciones sobre estos genes pueden llevar a ciertos tipos específicos de SHP y a causar alteraciones sobre algunos LRO (tabla 2) [38,39] (los genes que se encuentran entre paréntesis).
Tabla 2. Manifestación clínica y su LRO afectado
Diagnóstico
El diagnóstico de este síndrome se basa principalmente en las características clínicas encontradas en el paciente; la principal sospecha valorativa inicia en un individuo con albinismo oculocutáneo [7]. Según El-Chemaly [40], existen dos criterios diagnósticos que todo paciente con SHP debe presentar independientemente del subtipo que padezca: 1) albinismo oculocutáneo tirosinasa positivo y 2) un trastorno hemorrágico debido a una disfunción plaquetaria que varía de leve a grave.
Otro método de interés, que se relaciona con el segundo criterio, es el fenotipado plaquetario, el cual revela una ausencia o reducción significativa de los gránulos densos en las plaquetas. Esto puede demostrarse a través de la microscopía electrónica de montaje completo (EM). Una alternativa es confirmar la falta de una respuesta de agregación secundaria de las plaquetas ante estímulos exógenos a través de pruebas de agregación plaquetaria, lo que también apoya al diagnóstico del SHP. La cuantificación de la captación de mepacrina y el análisis de microscopía de inmunofluorescencia de alta resolución [41] son posibles métodos para diagnosticar la deficiencia de gránulos densos. La detección de variantes bialélicas en uno de los 11 genes asociados con el SHP mediante pruebas genéticas, como secuenciación de exones, secuenciación de Sanger, secuenciación completa de región codificante, secuenciación de panel por NGS, son los métodos más confiables para el diagnóstico de esta patología y, además, permite determinar el subtipo de SHP padecido por el paciente [3].
En pacientes que tienen alta sospecha de este trastorno, la determinación de la ausencia o disminución de gránulos densos en plaquetas con la ayuda de microscopía electrónica representa un pilar diagnóstico [41]. Posteriormente, se sugiere realizar un análisis genético; al respecto, NGS es una de las pruebas más confiables gracias a su capacidad para identificar variantes patogénicas bialélicas en los genes asociados con esta condición, pues detecta variantes en múltiples genes asociados al HPS al mismo tiempo, de manera más rápida y es menos costosa en relación con la secuenciación del exoma [42].
Tratamiento
Esta patología requiere un manejo integral (Tabla 2). Con el fin de proteger la vista de estos pacientes, se recomienda el uso de lentes y gafas que bloqueen los rayos UV, en caso de haber estrabismo puede ser corregido con cirugía. En el aspecto dermatológico, se recomienda vestir con prendas que protejan al tejido cutáneo para evitar el riesgo ya descrito de cáncer de piel. En caso de crisis hemorrágicas, se deben tener planes establecidos por si es necesario transfundir plaquetas o glóbulos rojos. Los individuos que padezcan neutropenias pueden estar sujetos a planes de prevención de infecciones y uso de estimuladores de colonias [5,7].
En el caso de las infecciones pulmonares y la fibrosis pulmonar, se debe prevenir el consumo de cualquier tipo de cigarrillo, además de brindar un tratamiento óptimo para el asma e infecciones pulmonares, y se aconseja la vacunación en estos individuos para reducir la incidencia de patologías respiratorias [7]. Es importante mencionar que no existe evidencia de que los corticoesteroides mejoren la fibrosis pulmonar en estos pacientes, únicamente la pirfenidona ha demostrado ligera eficacia [1,43]. La pirfenidona es una piridona que se ha empleado en el manejo de la fibrosis pulmonar por el SHP.
En un estudio publicado en la European Review for Medical and Pharmacological Sciences que evaluó a 13 247 pacientes con fibrosis pulmonar asociada a SHP tratados con pirfenidona, demostró que este fármaco contribuye a ralentizar la disminución de la capacidad vital forzada de estos pacientes, y a mejorar las distancias recorridas en la prueba de marcha de 6 minutos, menor mortalidad por cualquier causa y una reducción del riesgo de muerte por fibrosis pulmonar. Sin embargo, los pacientes expuestos a este fármaco tuvieron reacciones adversas relacionadas con el tracto gastrointestinal, la dermis, la función hepática y el sistema nervioso. A pesar de todo, sigue siendo una excelente opción para el manejo de esta enfermedad y es un medicamento bien tolerado [44].
La terapia génica y edición genética para la fibrosis pulmonar están siendo actualmente estudiadas en el SHP. Estas buscan restaurar la función de un gen codificador de proteína produciendo el aumento o la edición de un gen defectuoso a través de la administración de un gen completamente funcional empleando ADN, ARN u oligonucleótidos con vectores virales o no virales, sobre todo para el SHP1. Sin embargo, este tipo de tratamientos aún se encuentran en estudios preclínicos, considerando que aún existen varios desafíos para su adecuado funcionamiento, tales como encontrar una vía adecuada de administración, la selección de población de células diana adecuadas, la elección adecuada del momento de administración para prevenir o para el tratamiento de la enfermedad y la ausencia de un biomarcador capaz de medir específicamente la eficacia del tratamiento [45]. La terapia más viable para la fibrosis pulmonar por el SHP es el trasplante pulmonar bilateral o de un único pulmón [7].
En el caso de la colitis granulomatosa producida por esta enfermedad, los esteroides sí han mostrado respuesta; otras opciones incluyen fármacos antiinflamatorios, moduladores inmunitarios o fármacos antifactor de necrosis tumoral-alfa. Las opciones de tratamiento para pacientes que no responden a estas medidas son principalmente quirúrgicas, la colectomía parcial o total ha demostrado ser efectiva en estos casos [7]. En casos refractarios, fármacos como el rituximab y sobre todo el infliximab han sido ampliamente descritos y han demostrado su efectividad frente a medicamentos como corticoesteroides y azatioprina, que no han podido controlar la enfermedad [46]. No obstante, el infliximab no resulta útil en todos los casos de SHP, puesto que su efectividad en algunos casos es variable. Hay que considerar que una de las complicaciones de este fármaco es la reactivación de infecciones granulomatosas latentes [46].
En conclusión, el tratamiento del SHP incluye un sinnúmero de medidas tanto de prevención como de tratamiento, que requieren un seguimiento continuo y el estudio de todas las opciones de manejo, considerando cuál es la más viable en cada subtipo específico de SHP y en el caso de cada paciente.
Contexto poblacional
Si bien el síndrome de Hermansky-Pudlak es una enfermedad genética rara, en Puerto Rico es generalmente muy frecuente; se estima que, de 1800 personas vivas de ascendencia puertorriqueña, una de ellas padecerá este síndrome. Sin embargo, a pesar de ser una patología frecuente en este país, dicha comunidad sigue enfrentando desafíos en el manejo de esta enfermedad [6].
Los problemas que afectan el manejo de esta enfermedad son especialmente las limitaciones en el acceso a diagnósticos precisos y tempranos, aparte de la escasez de tratamientos especializados y recursos óptimos para el cuidado de los pacientes. Un problema grave en este país ha sido la exclusión de las personas que padecen de este síndrome al intentar recibir un trasplante pulmonar, a pesar de que la fibrosis pulmonar representa la principal causa de muerte por esta enfermedad. Este problema refleja la falta de recursos del país para enfrentar el manejo de esta complicación y, además, las desigualdades en el sistema de salud [47].
Otro problema lo representan las barreras educativas y de concienciación sobre el SHP en médicos y en la población en general, lo cual provoca retrasos en el diagnóstico de esta enfermedad y también una menor comprensión de todas las necesidades que requiere el manejo de estos pacientes, ya que, en ocasiones, ellos son quienes instruyen al médico acerca de su enfermedad. Otro factor adverso son los planes de prevención ineficientes en el tratamiento de estos pacientes los cuáles generan emergencias médicas fácilmente prevenibles si se tuviera una mejor instrucción acerca de la enfermedad. En Puerto Rico existen organizaciones como la Red del Síndrome de Hermansky-Pudlak, que se encarga de promover aspectos como la educación, apoyo e investigación para el progreso en el manejo de esta enfermedad [47].
Conclusiones
El síndrome de Hermansky-Pudlak (SHP) es una enfermedad autosómica recesiva que afecta múltiples sistemas, con 11 subtipos derivados de mutaciones en 11 genes asociados a cuatro subunidades proteicas (AP-3, BLOC-1, BLOC-2, BLOC-3). Descubierta por los médicos Frantisek Hermansky y Paulus Pudlak, la enfermedad se caracteriza por albinismo oculocutáneo, disfunción plaquetaria, colitis granulomatosa y fibrosis pulmonar (fibrosis pulmonar). La fibrosis pulmonar es la principal causa de muerte en estos pacientes, reduciendo su esperanza de vida a entre 30 y 50 años.
La prevalencia del SHP es baja, afectando a 1-9 personas por cada millón, con una mayor frecuencia en Puerto Rico. Los pacientes de este país principalmente presentan los subtipos SHP-1 y SHP-3.
Las manifestaciones clínicas incluyen alteraciones cutáneas (piel pálida, cabello claro), oculares (nistagmo, hipoplasia foveal) y diátesis hemorrágica (hematomas, epistaxis). La fibrosis pulmonar se presenta con disnea e hipoxemia progresiva, mientras que la colitis granulomatosa y la insuficiencia renal son complicaciones derivadas de la acumulación de lipofuscina ceroidea. La neutropenia e inmunodeficiencia también son comunes.
El diagnóstico se basa en características clínicas, análisis de gránulos plaquetarios y pruebas genéticas para identificar variantes patogénicas en los genes relacionados con SHP. El tratamiento es integral e incluye múltiples medidas que permitan prolongar la vida de estos individuos.
Financiamiento
Extendemos un agradecimiento cordial a la Universidad Técnica Particular de Loja.
Conflictos de interés
Los autores de este artículo no poseen ningún conflicto de interés.