
Mayra Delgado Cruz 1, Carlos Córdova Chacón 2, Cristina Córdova Tomalá 3, Santiago Bueno Lara 4
1 Especialista en Medicina Interna. Hospital Teodoro Maldonado Carbo IESS, Guayaquil, Ecuador
2 Médico General. Hospital Teodoro Maldonado Carbo IESS, Guayaquil, Ecuador
3 4 Estudiante Pregrado de Medicina. Universidad Católica Santiago de Guayaquil, Guayaquil, Ecuador
![]() 1. ORCID: 0000-0002-5472-0914
1. ORCID: 0000-0002-5472-0914![]() 2. ORCID: 0000-0002-8913-136X
2. ORCID: 0000-0002-8913-136X![]() 3. ORCID: 0009-0007-1138-8855
3. ORCID: 0009-0007-1138-8855![]() 4. ORCID: 0009-0007-2720-5451
4. ORCID: 0009-0007-2720-5451
Esta patología, también llamada complejo de esclerosis tuberosa (CET o TSC, por sus siglas en inglés), es una enfermedad genética multisistémica poco frecuente, que causa tumores benignos en el cerebro y en otros órganos vitales como los riñones, el corazón, los ojos, los pulmones y la piel. Es causada por defectos o mutaciones en dos genes, TSC1 y TSC2; solamente uno de los genes necesita ser afectado para que ocurra el CET. Aunque algunos individuos pueden heredar el trastorno de un padre que padezca de CET, la mayoría de los casos ocurren por mutaciones espontáneas (4). En nuestro caso, el padre del paciente fue diagnosticado de VHL, el cual fue heredado con una característica autosómica dominante. La clínica es variada dependiendo del órgano afecto y anormalidades de la piel característica específica. Los tumores pueden crecer en cualquier órgano, pero ocurren más comúnmente en el cerebro, los riñones, el corazón, los pulmones y la piel. Los quistes y angiomiolipomas renales ocurren entre aproximadamente un 40 a 80% de los individuos que padecen de CET, y se manifiestan por lo común entre las edades de 20 y 30 años; en su mayoría son pequeños, aparecen en números limitados y no causan ninguna complicación grave; su comportamiento es similar al de la enfermedad renal poliquística.
En nuestro paciente, la presencia de un quiste renal único sin afección renal nos hace dudar aún más de que se trate de CET. Los angiomiolipomas, las máculas hipomelánicas («manchas tipo hojas cenizas»), manchas o puntos rojizos llamados angiofibromas faciales y los fibromas ungueales o subungueales, que se encuentran en la esclerosis tuberosa y son características primordiales para la CET, no fueron encontradas en nuestro paciente. A nivel cerebral, los tubérculos corticales que se forman frecuentemente en la superficie de este órgano, pero también en las áreas profundas, los nódulos subependimales, que se forman en las paredes de los ventrículos; dichas características se visualizan en TC de cerebro o una resonancia magnética (MRI, por sus siglas en inglés), y no son hallazgos encontrados en el paciente por lo cual se descarta este diagnóstico.
Neoplasia endocrina múltiple
Las neoplasias endocrinas múltiples (MEN) son síndromes de herencia autosómica dominante caracterizados por la asociación de lesiones en distintas glándulas. El síndrome MEN tipo 1 incluye hiperparatiroidismo primario, tumores pancreáticos y adenomas hipofisarios; con menos frecuencia pueden aparecer adenomas suprarrenales, tumores tímicos y bronquiales, lipomas y varias lesiones cutáneas. Las manifestaciones clínicas incluyen con mayor frecuencia hiperparatiroidismo e hipercalcemia asintomática, que puede revelar nefrolitiasis; estos datos de laboratorio no se encontraban alterados en nuestro paciente. Los tumores de las células de los islotes pancreáticos se presentan en el 30 al 90% de los pacientes con MEN tipo 1, suelen ser multicéntricos y en ocasiones sintetizan varias hormonas.
Otros tumores enteropancreáticos funcionantes en la MEN 1 son los tumores de células no beta, que pueden ocasionar una diarrea secretora grave con depleción hidroelectrolítica, y a menudo provocan hipersecreción de glucagón, somatostatina, cromogranina o calcitonina, secreción ectópica de ACTH (hormona adrenocorticotrópica) o de hormona liberadora de corticotropina (responsable del síndrome de Cushing) e hipersecreción de hormona liberadora de hormona de crecimiento (responsable del desarrollo de acromegalia) (5).
Todas estas manifestaciones del síndrome MEN tipo 1 se descartan en el caso actual, pues nuestro paciente presentaba en TC de abdomen múltiples imágenes quísticas simples a nivel pancreático sin afectación endocrina alguna.
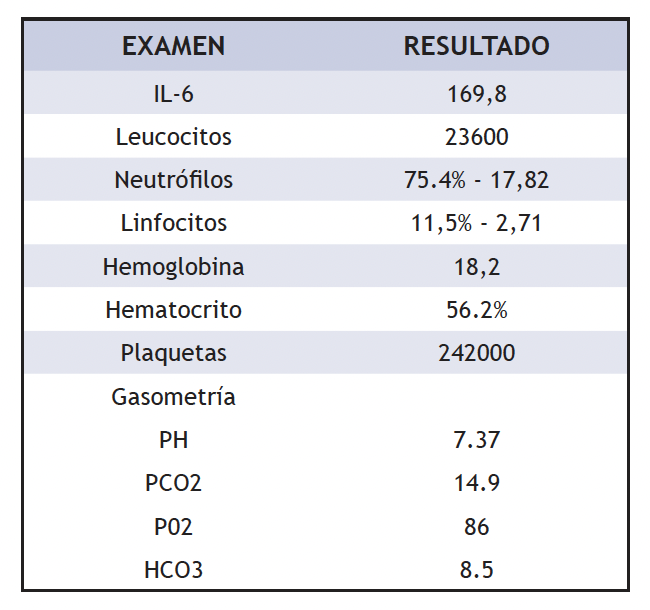
Sindrome Von Hippel-Lindau La enfermedad de Von Hippe-Lindau (VHL) es un síndrome neoplásico autosómico-dominante, con manifestaciones en múltiples órganos, provocado por la mutación del gen supresor de tumor del brazo corto del cromosoma 3 en la posición 3p25-26, caracterizado por la aparición de tumores benignos y malignos que afectan el sistema nervioso central, riñones, páncreas, glándulas adrenales y los paraganglios (6) (7). El gen VHL normal es un gen de supresión tumoral, es decir, su función a nivel de tejidos u órganos es la de suprimir o prevenir la formación de tumores. El síndrome de VHL no presenta un único síntoma principal, pues puede afectar a varios órganos a la vez, y la edad de aparición varía enormemente; la enfermedad es hereditaria, como en este caso donde el padre fue diagnosticado de VHL. Por otro lado, la aparición y la gravedad de la enfermedad puede variar mucho y existir dentro de una misma familia individuos que están afectados levemente, mientras que otros pueden tener manifestaciones graves (8) (9). La clasificación según el genotipo-fenotipo es la siguiente (10):
Elaborado: Dra. Angelita Fierro
En este caso, el paciente cumple de manera parcial los criterios de clasificación VHL tipo 1, ya que presentó angioma de la retina, hemangioblastomas en SNC, quistes renales y pancreáticos; sin embargo, el cáncer renal fue la excepción. Estas variaciones han sido evidentes en varios estudios que indican que la aparición de los tumores es posible a lo largo de la vida. Además, estas variaciones se desarrollan el grupo étnico al que pertenece el individuo; por ejemplo, las familias francesas suelen presentar lesiones del SNC; las alemanas, feocromocitomas; y las japonesas, tumores renales predominantemente (10).
Las manifestaciones clínicas se presentan de acuerdo al órgano afecto: La angiomatosis de la retina puede causar desprendimientos, hemorragias y, eventualmente, pérdida de la visión (11) (12). Los hemangioblastomas del SNC se manifiestan con cefalea, inestabilidad de la marcha, vómitos, alteraciones del equilibrio, así como debilidad de las extremidades. Las feocromocitomas pueden ser asintomáticos (13) o producir una gran variedad de síntomas: los más frecuentes son cefalea, sudoración, palpitaciones con taquicardia o sin ella, nerviosismo, pérdida de peso e hipertensión arterial. Otras señales incluyen la hipotensión ortostática, hiperhidrosis, arritmias, disnea, parestesia, polidipsia, mareos, crisis de epilepsia tipo gran mal, palidez, bradicardia, hematuria indolora, disartria y el temblor (9). En el caso que nos ocupa, el paciente muestra sintomatología predominantemente neurológica al momento de su llegada.
El tratamiento es la prevención de complicaciones, como el crecimiento tumoral al que está predispuesto el paciente, mediante el seguimiento periódico y multidisciplinario, puesto que, en el 2016, fue sometido a exéresis de hemangioblastoma cerebeloso y bajo supervisión oftalmológica por el angioma de retina. Abandonó su seguimiento cerca de 3 años y, al ingreso al Hospital Teodoro Maldonado Carbo, se evidenció un nuevo hemangioblastoma cerebeloso y múltiples imágenes quísticas a nivel pancreático, además de gran quiste renal derecho que requería de una nueva resección quirúrgica, y completar con tratamiento médico por la pérdida de la visión o los trastornos neurológicos asociados.
Los autores declaran que no existe conflicto de intereses.
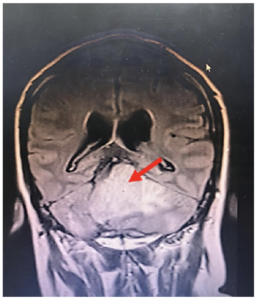
Figura 1. Hemangioblastoma en fosa cerebral posterior
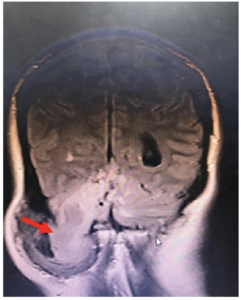
Figura 2. Fístula de LCR, por procedimiento previo
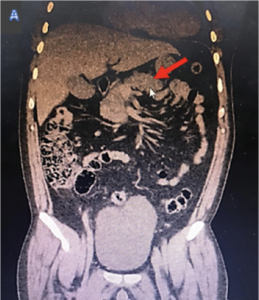
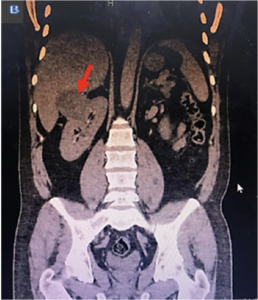
Figura 3. A. TAC de abdomen. Múltiples imágenes quísticas en páncreas. B. Imagen quística renal derecha de gran tamaño